

Where AI is actually changing pharma: four workflows that are already producing results

Originally published July 2024 in PharmaPhorum and Pharma Compliance Monitor.
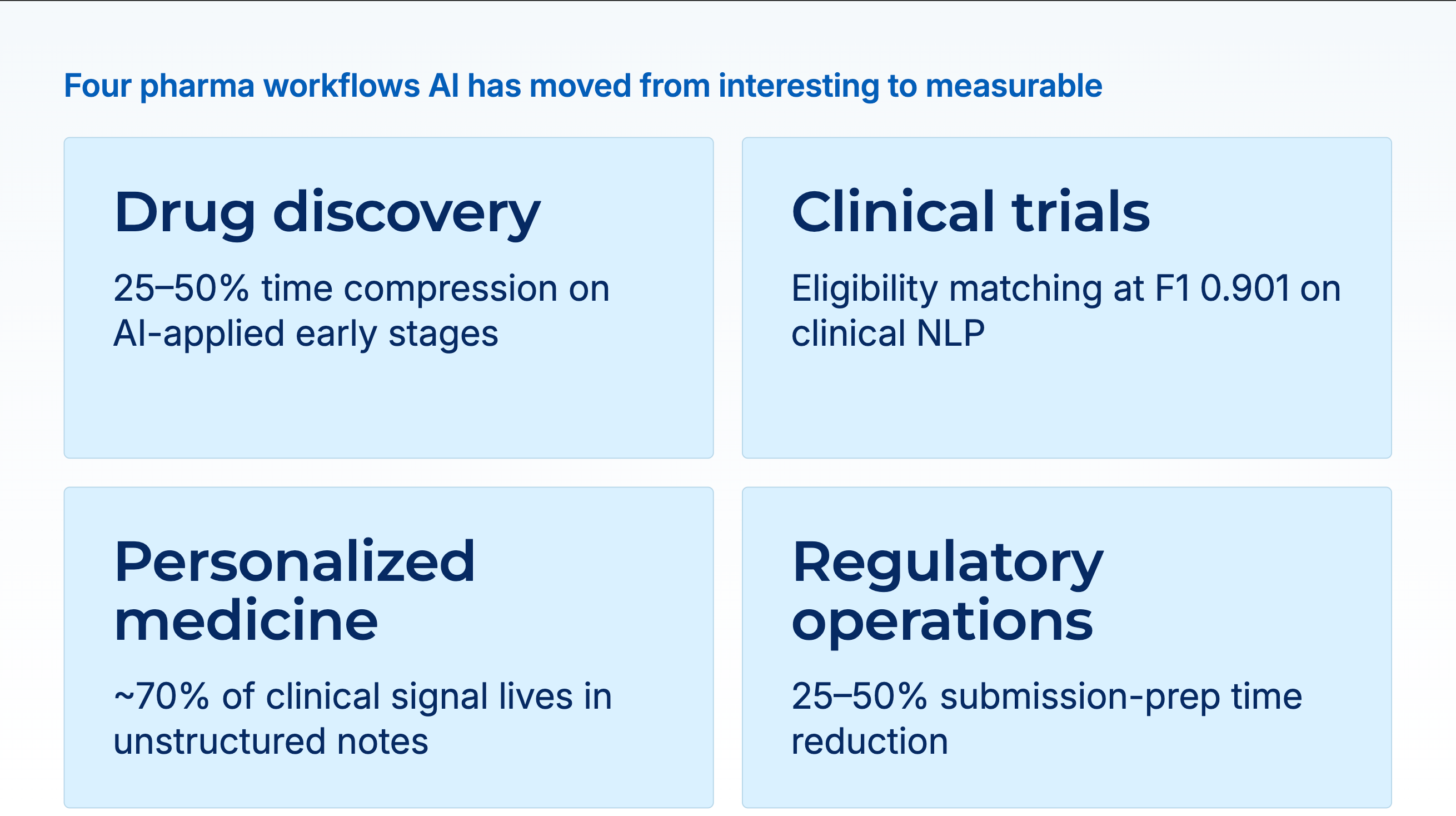
Pharma has spent a decade piloting AI and a year arguing about generative AI. The useful question for an R&D leader or a chief compliance officer in 2024 is narrower than the pilot deck: which specific workflows have AI moved from interesting to measurable, and what does a pragmatic investment look like in each? Four areas fit that description today: earlier-stage target and lead identification, clinical trial design and patient recruitment, real-world-evidence-driven personalization, and regulatory and compliance operations. None of them is science fiction. All four have production case studies, peer-reviewed evaluations, and real cost and timeline savings attached. The rest is execution.
Drug discovery: AI has compressed the early funnel
The traditional drug-development timeline is 12 to 15 years from target identification to approval, at an average cost around $2.5 billion per approved drug. Most of that money is spent on candidates that eventually fail. AI cannot change the biology, but it can change the economics of the early funnel: where you generate candidates, score them, and decide which ones to move forward.
Three capabilities have matured to the point of being operationally useful. The first is structure-based candidate generation: generative chemistry models that propose small molecules matched to a target’s binding site, filtered by predicted ADMET properties. The second is virtual screening: computational evaluation of millions of compounds against a target, yielding a shortlist that chemists actually test. The third is genomic and multi-omic target identification: models that mine genetic, proteomic, and phenotypic data to propose targets associated with a disease, or to identify why a specific patient population responds to a therapy and another does not.
The evidence for time savings is stacking up. Companies leveraging AI in early discovery have reported development-time reductions of 25% to 50% on the stages where AI is applied. Insilico Medicine moved an AI-designed drug candidate through discovery and preclinical stages in roughly 30 months, a cycle that historically ran 4 to 6 years. The 2024 AI-aided drug-discovery pipeline expanded at roughly 40% year-over-year growth.
What hasn’t changed, and what pharma leaders should be careful not to oversell internally, is the back half of the funnel. Phase 2 and Phase 3 failures still happen, and AI-designed drugs are not immune. The 2023 failure of ulotaront, an AI-aided TAAR1 agonist for schizophrenia, in its Phase 3 studies is a useful counterweight to the discovery-stage success stories. AI improves the hit rate in early filtering. It does not eliminate biological uncertainty in humans.
The practical investment pattern that works: fund AI platforms that integrate with your existing chemistry and biology workflows rather than standalone AI discovery tools; require every predicted property to have a confidence interval and a data lineage that your med chem team can interrogate; and track reduction in candidates-screened-per-hit as the operational KPI, not “number of AI-designed drugs in pipeline.”
Clinical trials: patient recruitment and protocol design are the bottlenecks AI actually moves
Clinical trials are where AI has the most immediate operational impact on pharma economics. Trials are slow, and most of the slowness is in two places: finding patients who meet protocol eligibility criteria, and writing protocols that are tight enough to produce a clear answer without being so narrow that recruitment stalls.
Patient recruitment is a natural-language-processing problem. Eligibility criteria — “newly diagnosed, HER2-positive, no prior trastuzumab exposure, ECOG 0–1, adequate hepatic function” — need to be matched against each prospective patient’s entire medical history, most of which sits in unstructured clinical notes, pathology reports, radiology reports, and lab results rather than in structured EHR fields. Matching those criteria reliably requires clinical NLP that extracts entities, assertion status (is the condition present, absent, possible, or historical?), relations (which medication was given for which condition?), and terminology-normalized codes (SNOMED, ICD-10, RxNorm) from the notes, then runs the eligibility logic on the structured result.
This is where healthcare-specific language models earn their cost. A 2024 *JAMIA* study on the 2010 i2b2 clinical-concept extraction benchmark measured GPT-4 at F1 0.804 with baseline prompting, against BioClinicalBERT, a 110-million-parameter domain-tuned model, at 0.901. The gap matters because a 10-point F1 drop on entity extraction cascades into false-positive and false-negative matches downstream. A trial that screens 10,000 patients and mis-matches 10% of them wastes months of coordinator time on chart reviews that should have been filtered out. Domain-specific models consistently outperform frontier LLMs on eligibility-relevant extraction tasks, and they do so at a fraction of the per-record cost, which is what makes population-scale screening economically feasible.
Protocol design is the other high-value AI application. Models trained on historical trial data can simulate enrollment rates under different eligibility criteria, stratify patient subgroups, and stress-test endpoints against real-world variability before the protocol is finalized. Bristol Myers Squibb has used machine-learning-based protocol optimization to accelerate patient recruitment and reduce costs. AstraZeneca has deployed AI-driven platforms for real-time monitoring of trial data, with measurable improvements in compliance tracking and decision turnaround. These are not pilot results, they are production operations at major sponsors.
The investment pattern: treat eligibility-criteria matching as a regulated NLP workflow, not as a feature of your EDC vendor’s dashboard. Demand domain-specific model benchmarks with peer-reviewed methodology. Require the system to run inside the health system’s environment or the sponsor’s environment, not in a third-party cloud, because the data involved is protected health information that most institutions will not release.
Personalized medicine: AI is what makes stratification operational
Personalized medicine has been a pharma talking point for 20 years. What changed recently is that the data infrastructure and the modeling capability are finally in place to operationalize the stratification logic at population scale.
The operational pattern: build a longitudinal patient record that combines structured EHR data (diagnoses, medications, labs), unstructured clinical notes (reasoning, symptoms, severity), genomic and multi-omic data where available, and patient-reported outcomes. Harmonize the combined record to a common data model (OMOP is the working standard for research and increasingly for pharma RWE). Train predictive models on the combined view to identify sub-populations that will respond to a therapy, sub-populations that will not, and sub-populations at higher risk of adverse events.
Two specifics that matter for the economics of this work. First, the majority of clinically relevant information about a patient lives in unstructured notes and reports, not in the coded fields. A personalization system that sees only structured data sees maybe 30% of the signal. Second, the extraction quality from unstructured sources is the binding constraint on downstream model quality. A cohort built from clinical NLP that runs at F1 0.90 on entity extraction produces materially different treatment-response predictions from one built from NLP that runs at 0.75, and the difference shows up as signal-to-noise in the predictive modeling downstream.
For pharma, the practical uses are consistent across therapy areas: responder and non-responder stratification on approved drugs; enrichment strategies for trial designs; post-approval patient-selection guidance via RWE studies; and biomarker discovery from multi-omic data paired with clinical outcomes. The largest measurable impact in 2024 is on trial enrichment, using RWE to identify which patient subtypes are most likely to respond to a mechanism of action, then designing the trial to enroll those subtypes preferentially. This shows up in both smaller-than-traditional trial sizes and in higher success probabilities.
Regulatory and compliance operations: the ROI story that rarely gets pitched at conferences
The area with the cleanest ROI and the least conference-stage coverage is regulatory and compliance operations. The work involved is unglamorous: labeling documents for submission, monitoring global guidance updates, reconciling internal quality events against external signals, preparing regulatory correspondence, tracking deviations and CAPAs, running pharmacovigilance case triage. It is also enormous, expensive, and highly rule-bound, which makes it exactly the shape of work AI is currently good at.
Three patterns have moved from pilot to production at large pharma:
Regulatory intelligence. Continuous monitoring of FDA, EMA, PMDA, and national-authority guidance updates, with automated identification of the ones that affect a specific product family. Gap analysis against the company’s own submissions and labels, surfacing the changes that require a response. The content is dense, multilingual, and fast-moving. Frontier LLMs do useful reasoning here once the source documents have been cleaned, classified, and indexed by domain-tuned NLP.
Submission-document preparation. Clinical Study Reports, Common Technical Documents, and similar submission artifacts involve compiling data from multiple sources, applying format and terminology conventions, and producing documents that must be internally consistent. AI assists with section drafting, cross-reference verification, terminology normalization, and consistency checking. The human authors are still responsible for the content; the AI removes the hours spent on coordination and formatting. Companies that have published numbers on this report development-time compression of 25% to 50% on submission-preparation stages.
Pharmacovigilance case triage. Adverse-event reports arrive in structured and unstructured form from clinicians, patients, call centers, and public sources. Most are routine; a minority contain safety signals that require urgent review. AI-based triage classifies cases by severity, extracts the relevant clinical entities, and routes high-signal cases to human reviewers while auto-processing the routine ones with human sampling for QA. This is the same human-in-the-loop architecture that works in clinical coding: calibrated AI running at high throughput, domain experts focused on the flagged cases.
The economics of compliance AI are attractive because the baseline is heavy manual work at high hourly rates. A 10% reduction in coordinator time across a global pharmacovigilance operation is a large number. A reduction in late-filing penalties from proactive guidance monitoring is a larger one. The risk profile is also favorable: these are internal workflows with human review in the loop, not patient-facing decision support, which means the deployment path is shorter than it is for clinical AI.
Making the investment decisions sort themselves
Four concrete questions for pharma leaders planning 2024 and 2025 AI spend:
Where is the binding constraint in each workflow you care about? For discovery, it’s usually the hit rate in the early funnel. For trials, it’s patient recruitment and protocol quality. For RWE, it’s data harmonization quality. For compliance, it’s coordinator throughput and consistency. AI investments aligned with the actual constraint produce measurable returns; investments that skip past the constraint to the shinier downstream step rarely do.
Does the vendor’s accuracy claim come with peer-reviewed methodology? “Our AI is 99% accurate” means nothing without the task, the dataset, the evaluation protocol, and the baseline. Production-grade pharma AI vendors publish their benchmarks in peer-reviewed venues and make their evaluation datasets available for customer reproduction. Vendors that do not should be discounted accordingly.
Where does the data live during processing? For any workflow touching clinical notes, patient records, or PHI, the answer is effectively required to be “inside the customer’s environment,” not “in the vendor’s cloud.” HIPAA, GDPR, and the patchwork of US state privacy laws have moved in-environment deployment from a premium feature to a procurement requirement.
Is there a human-in-the-loop layer designed as part of the system? For regulatory-grade workflows, calibrated AI routing uncertain cases to domain-expert reviewers is the architecture that hits the accuracy bars. Systems that skip this layer either over-promise on automation or under-deliver on throughput.
The headline for pharma leaders in 2024 is not that AI is transformational. It’s that AI has stopped being a slide in the strategy deck and started being a line item in the R&D and compliance budgets, because the workflows where it works have measurable outputs attached. The organizations moving from pilot to production on discovery-stage screening, patient recruitment, RWE-driven stratification, and compliance automation are getting meaningful timeline and cost reductions. The ones still debating whether to start are falling behind on a cycle that is no longer speculative.
FAQ
Is AI-aided drug discovery actually producing approved drugs, or just faster preclinical candidates?
As of 2024, AI has materially accelerated the early stages (target identification, lead generation, preclinical candidate selection) with documented 25% to 50% time compression on those stages. The first wave of AI-designed candidates is now in Phase 2 and Phase 3 trials. Success rates in clinical trials are biological questions that AI helps address via better patient stratification and trial design, not a problem AI solves by itself.
Why is clinical NLP such a large fraction of the AI work in trials?
Because eligibility criteria, adverse-event documentation, and the majority of clinically relevant patient information live in unstructured text, not structured EHR fields. Reliable trial operations require turning that text into structured data that downstream rules and models can act on. The quality of the NLP is the quality of the trial-operations layer on top of it.
What’s the realistic ROI timeline on AI for regulatory and pharmacovigilance operations?
Short, measured in months rather than years, because the baseline is expensive manual work and the human-in-the-loop architecture is well understood. Typical productive deployments show measurable coordinator-hour reductions within a quarter and move to broader rollouts within a year. This is usually the fastest ROI line in a pharma AI portfolio.
Can general-purpose frontier LLMs handle regulatory-document drafting?
They can assist on drafting and consistency checking once the input documents have been cleaned, classified, and indexed. They cannot be the whole pipeline, because submission-document preparation involves domain-specific terminology, cross-reference verification, and format conventions that reward specialized models. The production pattern is composition: domain-tuned models for the structured work, frontier LLMs for the drafting and summarization on top.
What’s the most common mistake pharma companies make when scaling an AI pilot to production?
Skipping the harmonization layer. A pilot on a curated, pre-cleaned dataset that reached 95% accuracy does not generalize to production on raw operational data: because production data is noisier, more variable, and more multilingual than the pilot set. The investment that makes the pilot generalize is the one in pre-processing, entity extraction, terminology normalization, and confidence calibration. Organizations that budget for the model but underfund the data layer routinely find their production accuracy 10–20 points below pilot accuracy.